How hard is it to model LK99 on a quantum computer?
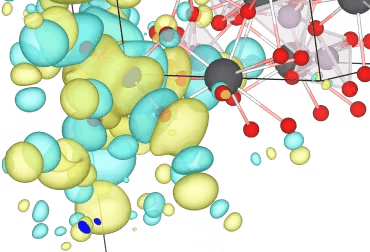
Wannier function of an active space band
The novel material LK99 has had the physics and materials science communities rushing in to verify and validate claims of room-temperature superconductivity. Attempts to theoretically simulate LK99 using industry-standard methods - density functional theory - produced inconclusive results and have highlighted the importance of accurately modelling quantum effects in materials. This is exactly the remit of quantum computers, and leads us to our titular question: how hard is it to model LK99 on a quantum computer? With the corollary question, when can we bring quantum computing to bear on such problems?
At Phasecraft we have answered this question using our proprietary material simulation approaches for encoding and compiling materials on quantum hardware. This is achieved by combining physically motivated arguments with algorithmic developments in our software tool "Magritte", the same approaches that have been used to build our materials quantum resource estimates detailed in our recent preprint [1].
Based on the insight gained during compilation of our quantum resource estimates, we expect that materials exhibiting a ‘flat-band’ structure will have favourable circuit depths and overall resource estimates. It so happens that LK99, and other related materials, have such a flat band structure, which gave additional motivation to understand if such materials could be early targets for quantum computation.
Following a classical/quantum hybrid workflow, we first establish a classical model of the LK99 structure that allows us to extract an approximate representation of the electrons within the material. From here we can identify and obtain a representation of the important electronic structure (the active space) responsible for potential superconductivity, and with this in-hand we can determine the encoding and quantify the required quantum hardware resources. We find a remarkable reduction of 8 orders of magnitude in complexity compared with previously developed methods.
LK99 Crystal Structure
LK99 has the chemical composition Pb9Cu(PO4)6O and is structurally derived from its parent lead-apatite compound Pb10(PO4)6O which is a wide gap insulator. It was reported to be a high temperature superconductor with the Cu-doping proposed as the origin of the superconducting mechanism. Most of the theoretical studies that have been reported since the original news of LK99 broke have treated both Pb9Cu(PO4)6O and Pb9Cu(PO4)6(OH)2. The structures of these two parent phases (i.e without Cu doping) are readily available in open access structure databases. It is reported a non-magnetic system without any coulomb correction can give the electronic flat-band [3]; for this reason we use the atomic structure for Pb9Cu(PO4)6O given in their supplementary material.
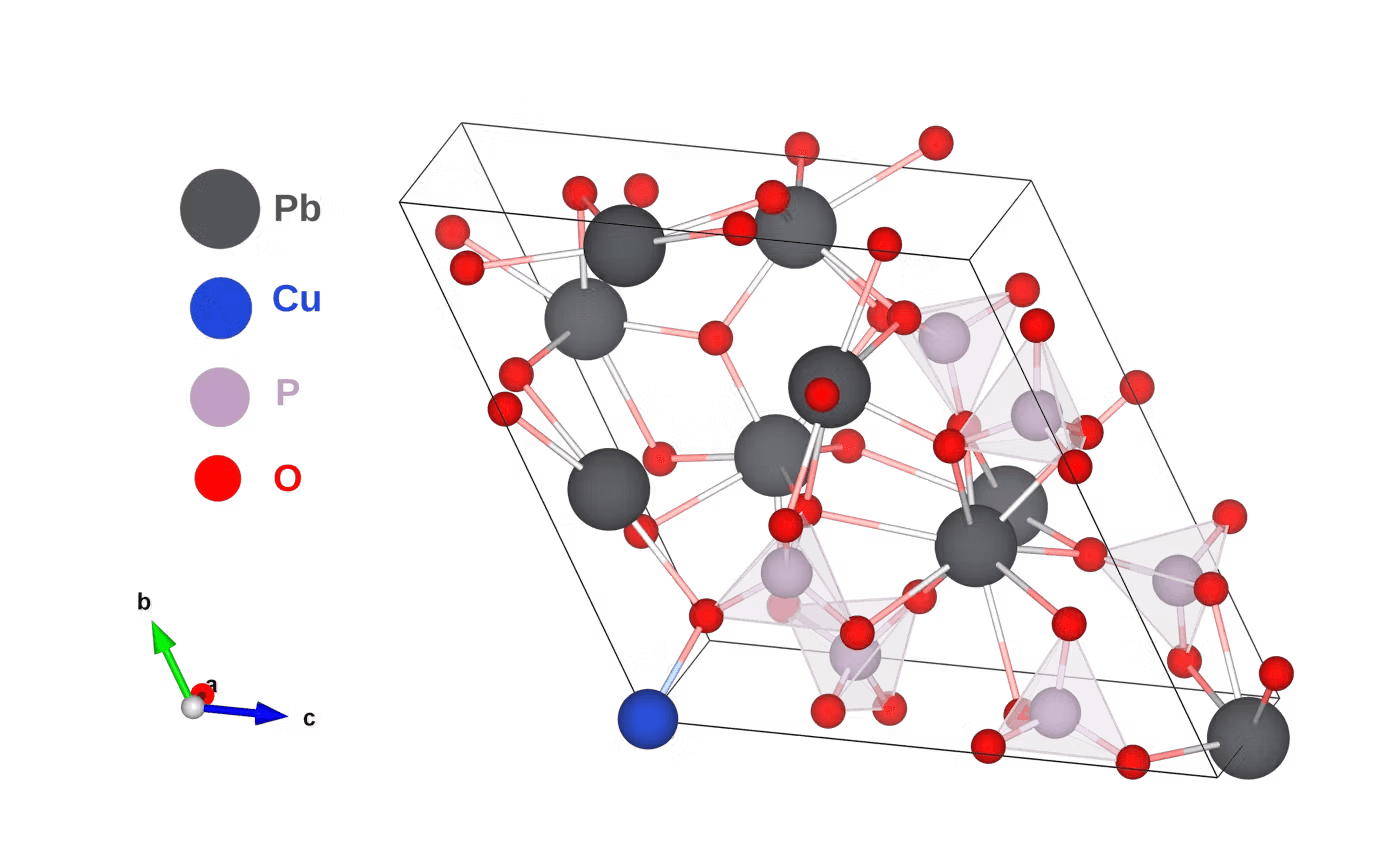
Figure 1: 3D representation of atomic structure used in this work.
After the structural relaxation we get the crystal structure detailed in the Table below (units of Angstrom) :
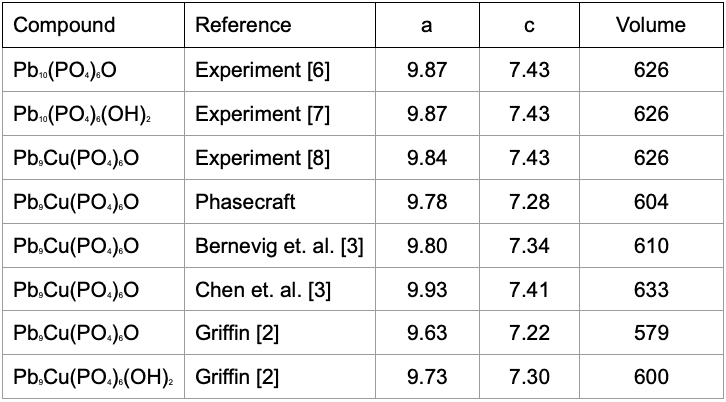
As we can see, compared with experiment (whose characterisation is not fully known) the DFT computations can vary (due to technical calculation differences), but are close to the reported experimental parameters. With this established we turn to the next step of considering the electronic structure.
LK99 Electronic Structure and Active Space Determination
Classical theoretical studies have observed an isolated set of 2 narrow Cu bands crossing the Fermi level, indicating that Cu doping induces a metal insulator transition [2,3,4,5], and potentially assists superconductivity. It has also been found that these systems favour ferromagnetism energetically but paramagnetism does not destroy the flat bands. At the time of writing despite the superconductivity claim being controversial [9], LK99 has nevertheless emerged as a material with interesting properties.
From the analysis shown in Figure 2 we can see that projecting onto Cu-d and O-p states within 1 eV of the Fermi level will describe a physically motivated active space. The two states at the Fermi level which fractionally occupy the Cu d9 level and have dyz and dxz characteristics, are chosen as the active space for the Wannierisation step.
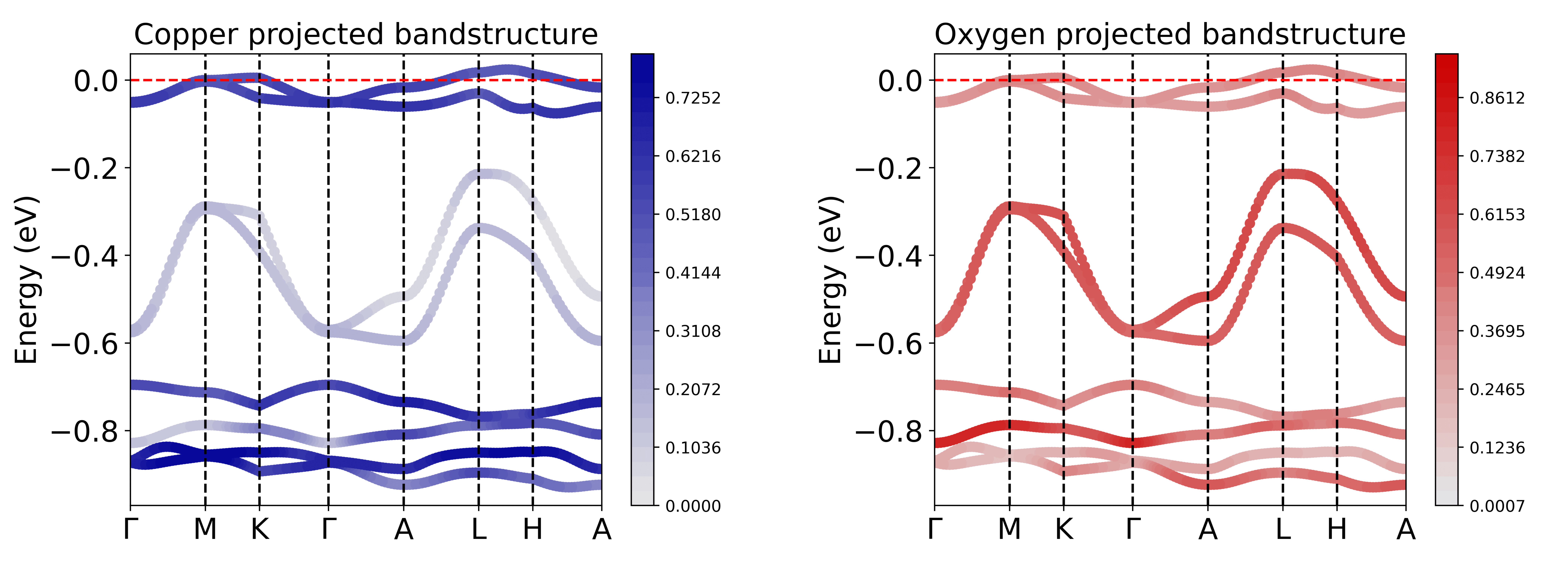
Figure 2: Contribution of atomic projectors on selected bands, left-hand side Cu-d, right-hand side O-p.
The Wannier functions are illustrated below in Figure 3, and are what get passed to our code Magritte. It can be seen in this case that the choice of bands gives a broad spread. Atomic character with a narrow spread of the functions can be retained by using larger active spaces. However, the aim here was to find the minimal space that could represent the essential physics of the system.
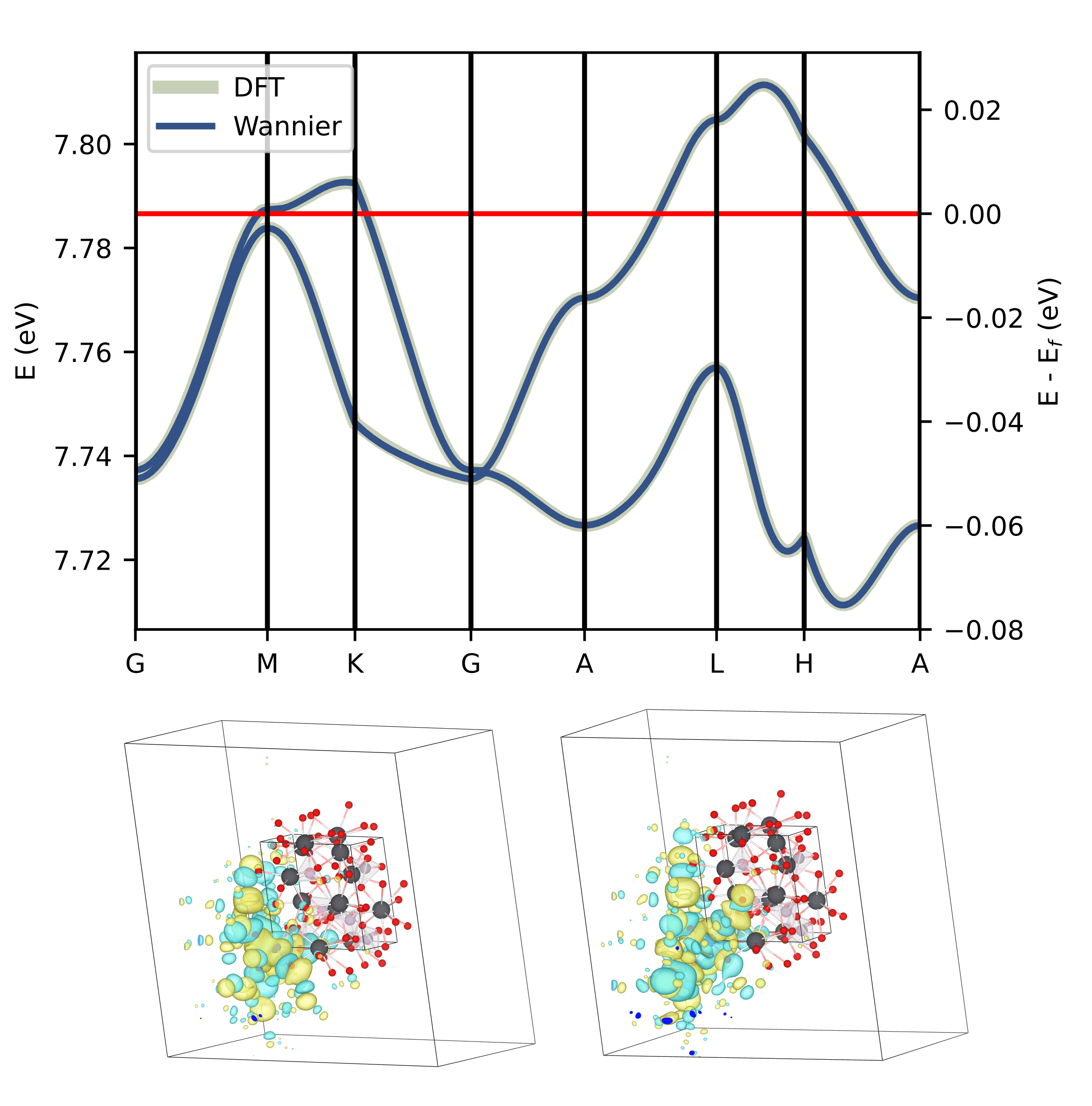
Figure 3: Band Structure of the active space, with real space image of Wannier functions obtained to describe these bands.
LK99 Quantum Resource Estimation
The next step in Magritte is to determine the size of motif (structural cell repeat) that is required to capture the periodic band structure in three dimensions. This is achieved by expanding out the number of cell neighbours until the band structure is reproduced within a defined tolerance. Here it was found that 11 neighbours gave us excellent agreement.
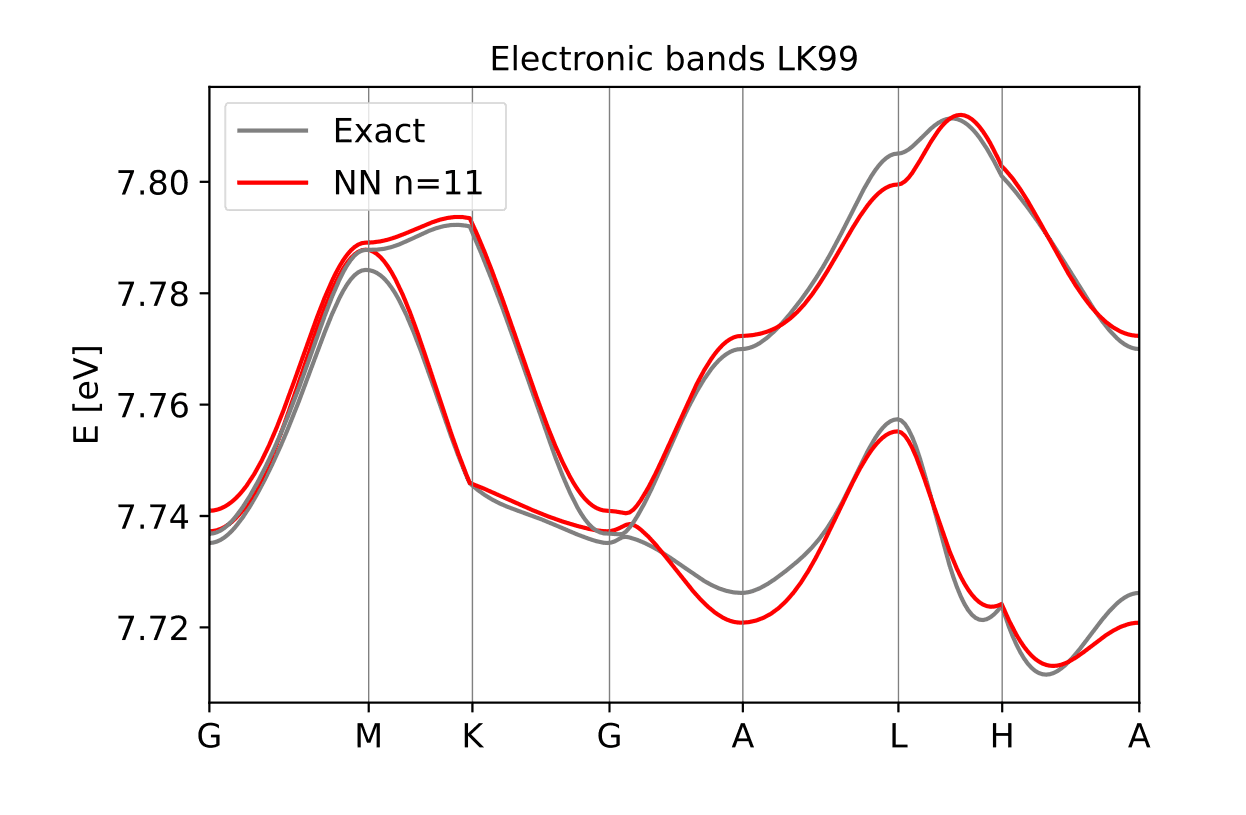
Figure 4: Band Structure obtained from Magritte for LK99, nearest neighbour (NN) for periodic motif.
We can then proceed to calculate the coulomb integrals from the hopping terms of our tight binding Hamiltonian. Through consideration of the size of hopping term, we are able to discard (set to zero) certain terms, to help reduce the problem, whilst maintaining numerical precision.
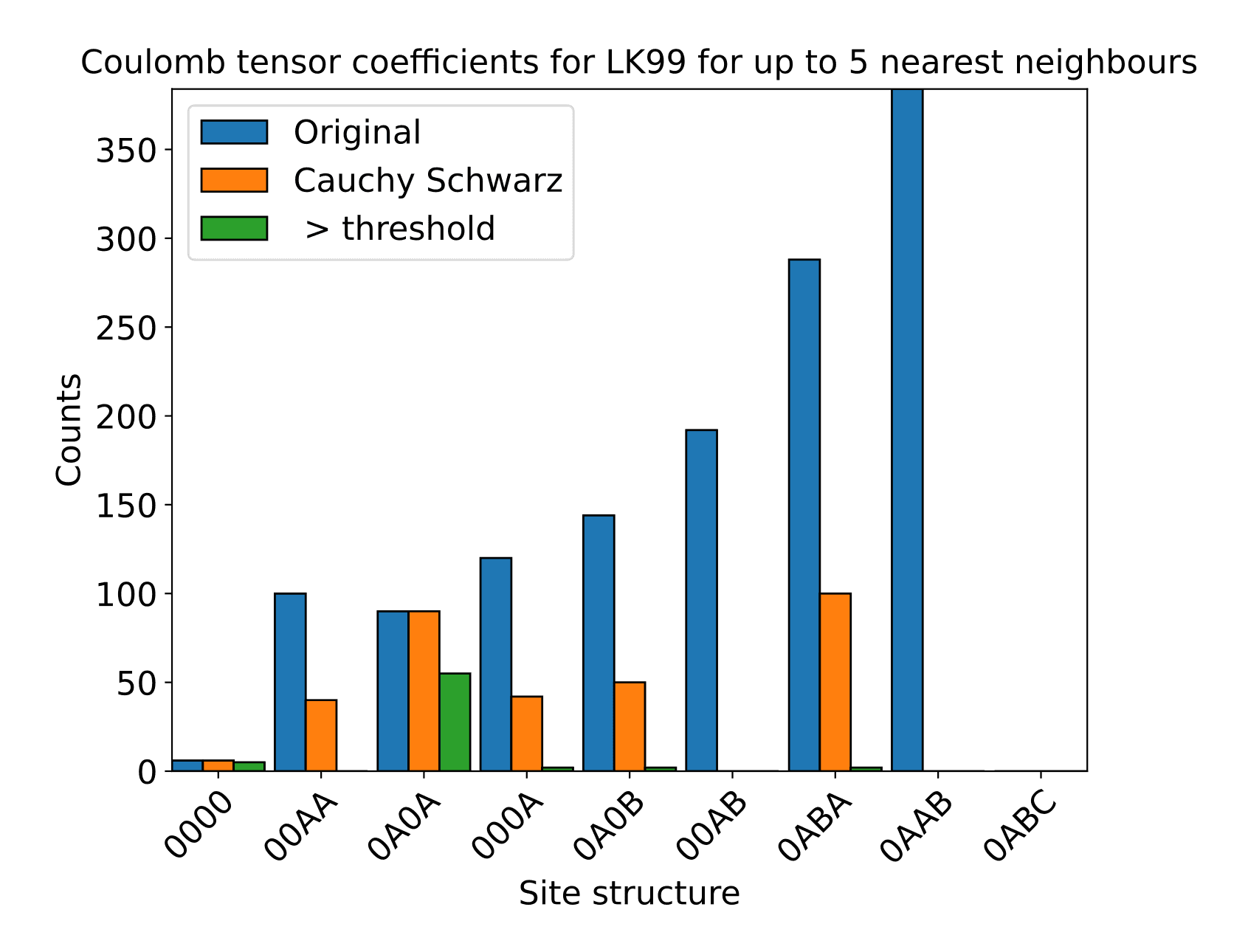
Figure 5: Number of interactions between periodic motif boxes, starting from original and following systematic reduction using the Cauchy Schwarz inequality and keeping only terms above a chosen threshold.
The resulting resources we obtain after considering the encoding and circuit construction are given in the Table below:
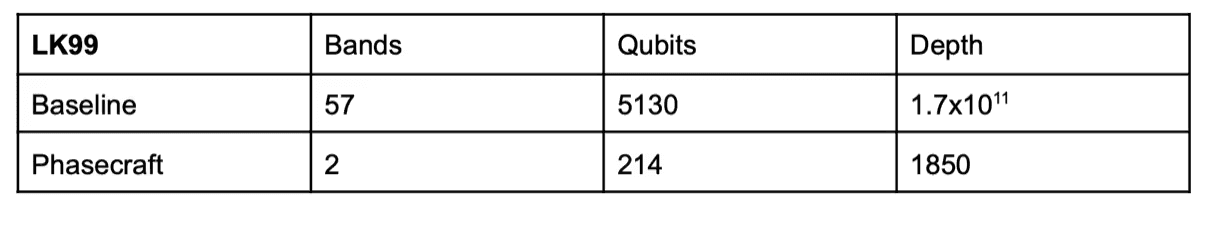
Here the depth corresponds to one layer of time-dynamics simulation or VQE with the Hamiltonian Variational ansatz. We note that whilst the narrow bands are not ‘molecular’ like, in the sense of having no inter-site hopping or interactions, the resource requirements are not so far away from our most favourable system studied to date (SrVO3, for which we also considered an active subspace). There is also room for improvement in the case of LK99, for instance; using a larger subspace would help localise the Wannier functions more, providing a shallower depth, but at the price of a higher qubit count.
Outlook
We have addressed our first question, how hard is it to model LK99 on a quantum computer? The answer being with the appropriate choice of active space, not so hard as to not be feasible in the near term. Regarding our corollary question of when can we bring quantum computers to bear on this problem, indications are maybe sooner rather than later. The qubit count is already within range of current devices. Gate fidelity is also a consideration, and the circuit depths produced are beyond the current state of the art by around an order of magnitude; but it can be seen that Phasecraft’s proprietary approach has reduced the problem by eight orders of magnitude, and there are some further optimisations we may be able to take to reduce this further. The electronic structure of this class of materials possessing relatively flat bands is favourable for hardware on manufacturers' near-term roadmaps and shows an opportunity to apply quantum computers to this challenging and current problem.
References
[1] Clinton et al. Towards near-term simulation of materials, https://arxiv.org/pdf/2205.15256.pdf
[2] Chen et. al. First-principles study on the electronic structure of Pb10−xCux(PO4)6O (x = 0, 1), https://doi.org/10.48550/arXiv.2307.16040
[3] Griffin, Origin of correlated isolated flat bands in copper-substituted lead phosphate apatite, https://doi.org/10.48550/arXiv.2307.16892
[4] Bernevig et. al. Pb9Cu(PO4)6(OH)2: Phonon bands, Localized Flat Band Magnetism, Models, and Chemical Analysis, https://doi.org/10.48550/arXiv.2308.05143
[5] Held et. al, Pb10-xCux(PO4)6O: a Mott or charge transfer insulator in need of further doping for (super)conductivity, https://doi.org/10.48550/arXiv.2308.04427
[6] Burns & Krivovichev, Crystal chemistry of lead oxide phosphates: crystal structures of Pb4O(PO4)2, Pb8O5(PO4)2 and Pb10(PO4)6O, Zeitschrift für Kristallographie - Crystalline Materials, 218(5), 357-365 (2009)
[7] Brukner et. al. Crystal structure of lead hydroxyapatite from powder X-ray diffraction data, Inorganic Chimica Acta, 236(1-2), 209-212 (1995)
[8] Kwon et. al. The First Room-Temperature Ambient-Pressure Superconductor, https://arxiv.org/abs/2307.12008
[9] Garrison, LK-99 isn't a superconductor - how science sleuths solved the mystery, Nature (2023)